



随着国内企业日益壮大,所以现在国内企业都将目光放到了海外市场。但想要将自己产品出口到海外市场有一个很大的问题,那么进入美国市场需要做什么认证呢?我们一起来了解一下。
1、什么是FDA认证
美国食品药品管理局(Food and Drug Administration 简称 FDA)的英文缩写,它是国际医疗审核权威机构,由美国国会即联邦政府授权,专门从事食品与药品管理的高执法机关。FDA是一个由医生、律师、微生物学家、药理学家、化学家和统计学家等专业人士组成的致力于保护、促进和提高国民健康的政府卫生管制的监控机构。通过FDA认证的食品、药品、化妆品和医疗器具对人体是确保安全而有效的。在美国等近百个国家,只有通过了FDA认可的材料、器械和技术才能进行商业化临床应用。
2、FDA覆盖产品范围
一、食品:所有出口到美国的食品(包括保健食品、饮用水、食品添加剂、婴幼儿食品等);
二、药品:药品原辅料、药包材、人类疫苗、处方药、非处方药等;
三、化妆品:护肤品、彩妆、洗发水等;
四、医疗器械:呼吸机、内窥镜、轮椅、电子体温计、血压计、口罩、防护服等;
五、激光辐射产品:微波炉、CT、X射线设备等;
六、食品接触材料:食物接触材料部件及原材料(各种塑料、金属、陶瓷、玻璃、竹木制品等);
七、兽医产品:牲畜饲料、宠物食品、兽药等。
3、FDA认证的作用
1.保护公众健康,确保食品是安全、健康、妥善标识的;确保药品、兽药、疫苗以及其他使用的生物制剂和医疗器械是安全和有效的
2.保护公众免受电子产品辐射
3.保证化妆品和膳食补充剂是安全的,并妥善标识
4.规范烟草制品
5.通过加快产品创新,促进公众健康
4、FDA认证的流程
1.产品归类,适合做检测规范引荐检测项目,适合注册规范的引荐做注册
2.填写检测或是注册相关申请表
3.需要做检测的需提供足够的样品到实验室进行测试
4.双方签订报价合同,安排付款
5.测试合格后发放合格报告,或注册证书
5、FDA认证怎么查询
只需登录FDA官网,输入相关编号信息即可查询信息
6、FDA注册有证书吗?
FDA注册没有相关证书,市面流传的FDA证书,是代理机构自己出的宣称性文件,是无效的,具体的可以从FDA官网下载相关注册文件及信息
7、FDA认证有效期
1.食品类偶数年有效(偶数年12月31到期)
2.化妆品永久有效
3.医疗产品每年12月31到期
8、FDA认证器械注册流程
(一)选择正确的路径递交
器械分类确定之后,需要选择相应法规要求下的上市前递交。常见的上市前递交类型包括:
- 510(k)(上市前通知)
- PMA(上市前批准)
- De Novo(自动III类指定的评价)
- HDE (人道主义器械豁免)
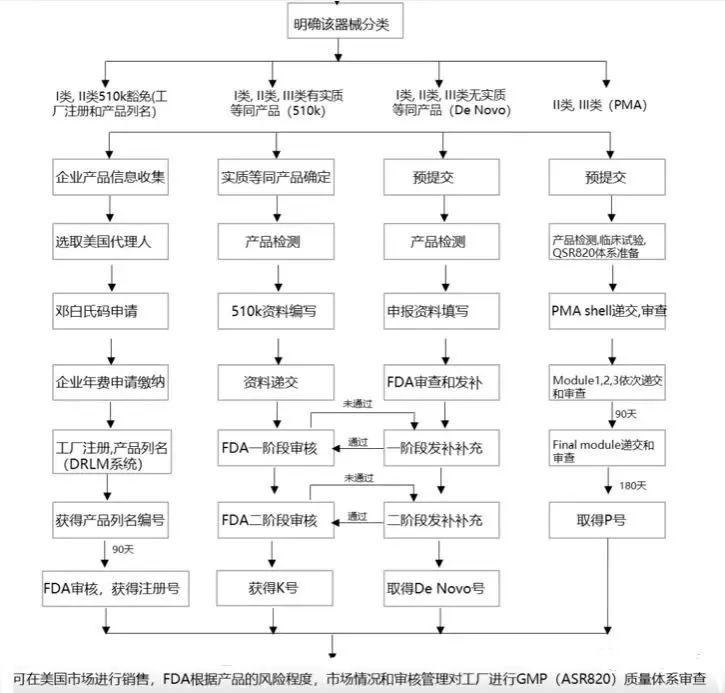
Ⅰ类以及大部分Ⅱ类器械要求以510(k)的方式递交。在510(k)递交过程中,申请者必须证明新的器械与对比器械在预期用途,技术特征以及性能测试方面实质等同。一些Ⅰ类和Ⅱ类器械可以豁免510(k),如果他们在21 CFR 862-892.9所述的豁免范围之内。这些豁免被列在21 CFR的分类规则中,也被汇集在医疗器械豁免文件中。
大部分Ⅲ类器械要求的递交方式为PMA。PMA为严格程度较高的上市前递交类型。在FDA批准PMA之前,申请者必须提供有效的科学证据,以证明器械预期用途的安全性以及有效性。
De Novo为没有有效对比的新器械提供一种方式,如果这种新器械满足特定标准,可以被分为Ⅰ或Ⅱ。
HDE为Ⅲ类器械提供了一种监管路径,这类器械预期对罕见疾病或状况的患者是有益的。器械有资格成为人道主义豁免器械,申请者必须获得人道主义使用器械(HUD)的指定,可通过向FDA的孤儿产品开发办公室Office of Orphan Products Development (OOPD)递交申请。
(二)准备材料
在选择正确的上市前递交类型之后,必须准备该递交类型所需的适当的资料。FDA开发一些帮助申请者准备上市前递交的资源类型,包括:
1.器械建议(Device Advice):—综合基于FDA上的网页法规协助
2.510(k)的准备:参考Premarket Notification 510(K)
3.PMA的准备:参考Premarket Approval (PMA)
4.CDRH学习(CDRH Learn):基于视频的教学模块,研讨会和录制的包括各种政策和指导力度的网络研讨会,CDRH递交前程序 —未来上市前提交申请可能要求FDA通过这个程序进行反馈。
准备上市前递交时需要考虑的信息:
(1)设计控制:所有II类以及III类器械根据质量管理体系(21 CFR 820.30)中对设计控制的要求进行设计。一些I类器械可豁免设计控制。
(2)非临床测试:器械上市要求的测试以及信息类型是通过器械的分类,作用机制,技术特征,以及标签来确定的。医疗器械上市前递交实施的非临床测试必须符合21 CFR 58中的良好试验管理规范(GLPs)
(3)临床证据:PMAs,HDEs 以及部分 510(k)s 和 De Novos 要求有临床证据。在早起的临床研究开始之前,研究申请者需要得到FDA器械临床研究豁免(IDE)的批准。这项研究也需要得到伦理审查委员会(IRB)的批准。临床研究必须符合所有的适用的器械临床研究豁免(IDE)法规以及良好试验管理规范(GLPs)。
(4)标签:器械的标签必须依据标签法规书写,且需要包含在上市前递交的资料中。
(三)递交资料
提交给FDA,并在FDA的工作人员审查过程中保持联系。
1.用户费用:在510(k)或PMA递交时,需要一定的用户费用
2.电子副本(eCopy):上市前递交必须包含以光盘(CD)、数字视频光盘(DVD),或闪存驱动器方式形成的电子副本。
3.行政备案审查:在上市前递交接收之后,FDA进行行政审查,评估递交是否是足够完整的,以接收实质性审查。
4.审查互动(Interactive Review):当递交的资料处于正在审查中时,FDA将和申请者保持联系以增加审查过程中的效率。
(四)完成登记
器械设备必须在FDA对其生产的企业进行登记,并对其器械进行列名。如果一个器械在上市前需要上市前清关(premarket clearance)或上市前批准(premarket approval),器械厂商在登记和列名之前必须等到它获得FDA的清关或批准。器械企业登记、登记号的分配或医疗器械的列名,都不意味着FDA对其企业或其产品的清关或批准。
免责声明:本文内容,图片来源于互联网及文摘转载整编而成,不代表本站观点,不承担相关法律责任。其著作权归其原作者所有。如发现本站有侵权/违法违规的内容,侵犯到您的权益,请联系站长,一经查实,本站将立刻处理。